Reportes de Casos
Raquitismo hipofosfatémico familiar ligado al cromosoma X: Reporte de Caso
Familial X-chromosome linked hypophosphatemic rickets: Report of a case
Raquitismo hipofosfatémico familiar ligado al cromosoma X: Reporte de Caso
Acta Médica Peruana, vol. 40, núm. 1, pp. 76-79, 2023
Colegio Médico del Perú

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial 4.0 Internacional.
Recepción: 19 Enero 2023
Aprobación: 22 Marzo 2023
Resumen: El Fósforo es regulado por el riñón y el sistema óseo orquestado principalmente por la acción de la parathormona (PTH) y una molécula recientemente descrita como el factor de crecimiento fibroblástico 23 (FGF-23) . Presentamos los casos de dos pacientes madre-hijo con Raquitismo hipofosfatémico ligado al cromosoma X. Se realizó el estudio genético identificándose una mutación en el Gen PHEX: variante patogénica tipo splicing en hemicigosis: mutación previamente descrita como HGMD CS126536. El Raquitismo Hipofosfatémico forma parte de un grupo de tubulopatías caracterizadas hiperfosfaturia. La mutación del gen PHEX con pérdida de función conduce al aumento de FGF-23. PHEX degrada el FGF-23 en fragmentos inactivos, evitando la excreción excesiva de fosfatos y el desarrollo de hipofosfatemia. En un paciente con hipofosfatemia no dependiente de la hormona PTH o de la vitamina D y de presentación familiar debe considerarse el diagnóstico de Raquitismo hipofosfatémico ligado al cromosoma X.
Palabras clave: Raquitismo, Hipofosfatemia, Cromosoma X.
Abstract: Phosphate is regulated by the kidneys and the osseus system, mainly due to the action of parathyroid hormone (PTH) and a recently described molecule, fibroblast growth factor 23 (FGF-23). We present the cases of two patients, mother and son with X-chromosome linked hypophosphatemic rickets. The genetic study was performed, and a mutation in the PHEX gene was identified, a splicing type pathogenic variant in hemizygosis. This mutation was previously described as HGMD CS126536. Hypophosphatemic rickets belongs to a group of tubulopathies characterized by hyperphosphaturia. PHEX gene mutation with function loss leads to increased FGF-23 levels. PHEX degrades FGF-23 into inactive fragments, preventing excessive phosphate excretion and the development of hypophosphatemia. In patients with PTH or vitamin D non- dependent hypophosphatemia, a diagnosis of X-chromosome linked hypophosphatemic rickets should be considered.
Keywords: Rickets, Hypophosphatemia, X Chromosome.
INTRODUCCIÓN
El fósforo (Pi) es uno de los cationes divalentes más importantes del organismo por la gran variedad de funciones que cumple: Es esencial para los procesos energéticos celulares, forma parte de la estructura de huesos, dientes, y es necesario en la estructura y función de la membrana celular. Este ion es regulado por el riñón y la parathormona (PTH) así como por una molécula identificada en el año 2000 como factor de crecimiento fibroblástico 23 (FGF- 23).[1-4] En el riñón la PTH modula la absorción y excreción de calcio y fósforo mientras que en el hueso se encarga del recambio de estos minerales según las necesidades del organismo.[4]
La concentración sérica de fosfato menor a 0,80 mmol/L en adultos se define como hipofosfatemia y menos de 0,30 mmol/L se clasifica como hipofosfatemia severa.[5] Existen tres mecanismos de hipofosfatemia: La disminución de la absorción intestinal, el aumento de las pérdidas urinarias y el paso desde el espacio extra al intracelular.[4] La hipofosfatemia de aparición en la infancia temprana se debe a problemas hereditarios y se caracteriza por la disminución de la reabsorción tubular renal de fosfato. Sus principales manifestaciones clínicas son el raquitismo y la osteomalacia.[6]
La forma más frecuente de la hipofosfatemia familiar es heredada como un rasgo dominante ligado al cromosoma X y se le conoce como raquitismo hipofosfatémico ligado al cromosoma X o hipofosfatemia ligada a X, esta alteración comprende casi 80 % de los casos de raquitismo hipofosfatémicos hereditarios.[6,7] La imposibilidad de inhibir el FGF-23 produce una incapacidad para la reabsorción tubular del Pi y reduce la actividad alfa- 1-hidroxilasa, lo que resulta en niveles séricos reducidos de 1.25-dihidroxivitamina D, hiperfosfaturia e hipofosfatemia.[8] Existen formas oligosintomáticas que se solo manifiestan algunos síntomas como dolor óseo o debilidad.
El raquitismo hipofosfatémico es una condición poco frecuente en nuestro medio y muchas veces difícil de reconocer en aquellos con signos o síntomas subclínicos por lo que el objetivo de este reporte de caso es presentar el espectro de presentación, diagnóstico y opciones de manejo de dos pacientes (madre-hijo) con raquitismo hipofosfatémico ligado al cromosoma X.[9]
PRESENTACIÓN DEL CASO
La madre de nacionalidad peruana-española tiene 36 años y diagnóstico de raquitismo hipofosfatémico desde los 2 años. Debutó con debilidad muscular, retardo del crecimiento y retraso en la aparición de los dientes. Con el tiempo desarrolló deformaciones en varo en extremidades inferiores que dificultaron la movilización afectando su calidad de vida. La madre recibió hasta los 15 años calcitriol 1.2 mcg/d y fósforo 1936 mg 2 tab cada 8 h, en la actualidad persiste con las malformaciones esqueléticas, presenta talla corta, dolor en articulación de cadera, rodilla y limitación parcial al movimiento. Fue evaluada por otorrinolaringología y no presentó alteraciones auditivas. La madre actualmente recibe como terapia calcitriol 1.5 mcg/ día y Solución de Joulie (ácido orto fosfórico 85% 5.45g fosfato disódico 18.75g) 60 ml cada 8 horas.
| Madre | Niño | |
| Cr | 0.68 mg/dl | 0.28 mg/dl |
| Na/K | 136/ 3.8 meq/L | 135/4.3 meq/L |
| Ca | 10.23 mg/dl | 9.14 mg/dl |
| Fósforo | 2.2 mg/dl | 2.47 mg/dl |
| FA | 170 mg/dl | 1259 mg/dl |
| PTH | 36.8 pg/ml | 32 pg/ml |
| Vit D | 14.10 mg/ml | 33.1 mg/ml |
| Ca/Cr | 0.06 | |
| P/Cr | 2.26 | |
| Calcio orina | 0.9 mg/kg/d (52.5 mg/d) | |
| Fosforo orina | 31 mg/kg/d (1758 mg/d) | |
| TRPi% | 70% | 77% |
El hijo fue evaluado por el antecedente materno de raquitismo hipofofosfatémico, tiene 15 meses de edad y recibe tratamiento con aporte de fósforo Solución de Joulie 7 ml 4 veces/día (aporte de fósforo 52 mg/kg/d) y calcitriol 1 ug/día (0.5 ug C/12 horas) desde los 4 meses. A pesar del tratamiento y de tener una curva de crecimiento adecuado para edad tiene un retraso en la aparición de dientes y deformaciones en extremidades inferiores como se aprecian en las imágenes 1 y 2. Los exámenes auxiliares de la madre y el hijo al momento de la evaluación clínica inicial se muestran en la tabla 1.
En el caso de la madre se pudo hacer la colecta de orina de 24 horas, en el caso del hijo se utilizó la relación de electrolitos y creatinina en orina de una muestra aislada por la dificultad en recolectar orina de 24 horas y donde la hiperfosfaturia expresada en la baja tasa de reabsorción de fósforo en ambos pacientes es el hallazgo más representativo.
La madre fue sometida a un estudio genético en España para identificar la mutación puntual, dando como resultado: Mutación del Gen PHEX, variante patogénica tipo Splicing en hemicigosis: rs886041839:g.22190443 G>A Mutación previamente descrita como HGMD CS126536 confirmando el diagnóstico materno de raquitismo hipofosfatémico ligado al cromosoma X.
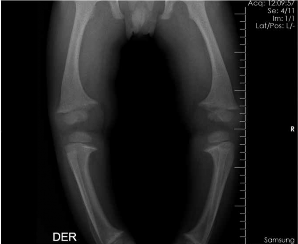
Figura 1.
Deformaciones en extremidades inferiores (Observación directa)
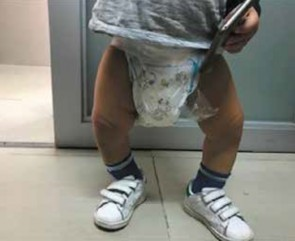
Figura 2.
Deformaciones en extremidades inferiores Rayos X
DISCUSIÓN
El raquitismo hipofosfatémico forma parte de un grupo de tubulopatías hereditarias caracterizadas por la pérdida excesiva de fósforo a través del riñón como defecto primario y donde el raquitismo hipofosfatémico ligado al cromosoma X (RHLX) tiene una incidencia de aproximadamente 1:20000 representando el 80 % de raquitismos hipofosfatémicos. Es la forma heredada más común de raquitismo en niños con 86-87 % de casos de herencia familiar y 57-72 % de casos esporádicos.[9,10]
La mutación del gen PHEX con pérdida de función (deleción del extremo 3’) conduce al aumento de FGF-23. El FGF-23 inhibe la transcripción de la 1 alfa hidroxilasa y es además una hormona fosfatúrica que requiere la presencia del cofactor Klotho. Las endopeptidasas PHEX (del inglés Phosphate regulating gene with Homologies to Endopeptidases on the X chromosome) y DMP1 controlan los niveles sanguíneos de FGF-23.[8]
En condiciones normales, PHEX degrada el FGF-23 en fragmentos inactivos, evitando de esta manera el aumento excesivo de la excreción de fosfatos y el desarrollo de hipofosfatemia. Sin embargo, las mutaciones en PHEX en los pacientes con RHLX permiten que se mantengan niveles elevados del FGF-23, esto induce alteraciones de la reabsorción tubular renal de fosfatos y desarrollo de hipofosfatemia.[9] Las manifestaciones clínicas ocurren después del primer año de vida con retraso en el crecimiento, raquitismo y deformidades óseas: Piernas curvadas, manifestaciones neurológicas, malformación de Arnold Chiari, craneosinostosis, sordera neurosensorial, tinnitus, vértigo.[10,11] Otras manifestaciones incluyen: abscesos dentales, artritis y calcificaciones de los tendones y ligamentos (entesopatía), estenosis del canal espinal y artropatía degenerativa.[11]
Se ha observado nefrocalcinosis entre el 22 % y 100 % de los pacientes con XLH. Esta variación se puede atribuir parcialmente a los estudios de pequeño tamaño y a la alta variabilidad de pacientes y tratamientos. En el caso de los pacientes aquí presentados ninguno desarrollo nefrocalcinosis ni litiasis renal.[10]
Los hallazgos de laboratorio más frecuentes incluyen: hiperfosfaturia, hipofosfatemia, calcio en sangre normal, calcio en orina bajo, 25 OH Vitamina D normal, 1,25 OH Vitamina D normal o baja, Fosfatasa alcalina alta, PTH normal o alta y FGF-23 alta.[11]
La hipofosfatemia grave induce el raquitismo y compromete la apoptosis de los condrocitos hipertróficos en la placa de crecimiento del hueso, generando las malformaciones óseas características de esta condición.[9-11]
El tratamiento se basa en la suplementación de fósforo: 40-100 mg/kg/día, calcitriol 15-60 ng/kg/día y últimamente Burosumab (inmunoglobulina humana recombinante que antagoniza los efectos y FGF-23). Esta última sustancia muestra resultados prometedores en la presente condición pudiendo ser un tratamiento seguro y clínicamente útil de los pacientes con enfermedades hipofosfatémicas relacionadas con el FGF23. Este hecho sentaría las bases para el establecimiento de una terapia única.[11]
En un paciente con hipofosfatemia no dependiente de la hormona PTH o de la vitamina D y de presentación familiar debe considerarse el diagnóstico de raquitismo hipofosfatémico ligado al cromosoma X.
REFERENCIAS BIBLIOGRÁFICAS
1. Anderson JJ. Calcium, phosphorus and human bone development. J Nutr. 1996;126(4 Suppl):1153S-8S. doi: 10.1093/jn/126. suppl_4.1153S.
2. Berndt T, Kumar R. Novel mechanisms in the regulation of phosphorus homeostasis. Physiology (Bethesda). 2009 Feb; 24:17- 25. doi: 10.1152/physiol.00034.2008.
3. Marks J, Debnam ES, Unwin RJ. Phosphate homeostasis and the renal-gastrointestinal axis. Am J Physiol Renal Physiol. 2010; Aug; 299(2):F285-96. doi: 10.1152/ajprenal.00508.2009.
4. Bergwitz C, Jüppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010; 61:91-104. doi: 10.1146/annurev.med.051308.111339.
5. Murer H, Forster I, Biber J. The sodium phosphate cotransporter family SLC34. Pflugers Arch. 2004; Feb; 447(5):763-7. doi: 10.1007/ s00424-003-1072-5.
6. Forster IC, Hernando N, Biber J, Murer H. Proximal tubular handling of phosphate: A molecular perspective. Kidney Int. 2006; Nov; 70(9):1548-59. doi: 10.1038/sj.ki.5001813.
7. Ariceta G, Langman CB. Growth in X-linked hypophosphatemic rickets. Eur J Pediatr. 2007 Apr; 166(4):303-9. doi: 10.1007/s00431- 006-0357-z.
8. Liu S, Gupta A, Quarles LD. Emerging role of fibroblast growth factor 23 in a bone-kidney axis regulating systemic phosphate homeostasis and extracellular matrix mineralization. Curr Opin Nephrol Hypertens. 2007 Jul; 16(4):329-35. doi: 10.1097/ MNH.0b013e3281ca6ffd.
9. Kinoshita Y, Fukumoto S. X-Linked Hypophosphatemia and FGF23- Related Hypophosphatemic Diseases: Prospect for New Treatment. Endocr Rev. 2018 Jun 1; 39(3):274-291. doi: 10.1210/er.2017-00220.
10. Beck-Nielsen SS, Mughal Z, Haffner D, Nilsson O, Levtchenko E, Ariceta G, de Lucas Collantes C, Schnabel D, Jandhyala R, Mäkitie O. FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J Rare Dis. 2019 Feb 26; 14(1):58. doi: 10.1186/s13023- 019-1014-8.
11. Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, Wicart P, Bockenhauer D, Santos F, Levtchenko E, Harvengt P, Kirchhoff M, Di Rocco F, Chaussain C, Brandi ML, Savendahl L, Briot K, Kamenicky P, Rejnmark L, Linglart A. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019 Jul; 15(7):435-455. doi: 10.1038/s41581-019-0152-5.